CG-seq: Comparative Genomics-seq
CG-seq is a software pipeline to identify noncoding RNAs in a genomic sequence by comparative analysis and multispecies comparison. It takes as input a genomic sequence (called the target sequence) and a set of other sequences coming from a variety of species to be compared against the target sequences.

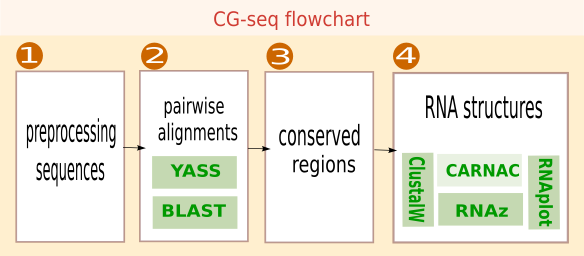
The algorithm of CG-seq proceeds in four steps.
- Preprocessing. Sequences are preprocessed to mask CDSs, or to remove redundancy between strains coming from the same species (optional).
- Alignment. The target sequence is compared to all other sequences to detect similar sequences across species.
- Conserved regions. Pairwise alignments are combined into clusters of significantly conserved regions.
- RNA structure. Conserved regions are investigated by inspection of evolutionary patterns to select sequences exhibiting a conserved consensus secondary structures.
Downloading and installation
![]() Linux, Mac OS X: CG-seq_linux.tar.gz. CG-seq is distributed under the GPL license.
Linux, Mac OS X: CG-seq_linux.tar.gz. CG-seq is distributed under the GPL license.
Documentation
CG-seq full documentation is available here.